## 9.4 現代環境下的 NHST:多重測試
到目前為止,我們已經討論了一些例子,其中我們對檢驗一個單一的統計假設感興趣,這與傳統科學是一致的,后者通常一次只測量幾個變量。然而,在現代科學中,我們通常可以測量每個個體數百萬個變量。例如,在量化整個基因組的基因研究中,每個個體可能有數百萬個測量單位,在大腦成像中,我們常常同時從大腦中超過 100000 個位置收集數據。當標準假設檢驗應用于這些環境中時,除非我們采取適當的措施,否則會發生不好的事情。
讓我們看一個例子來看看這是如何工作的。理解能使個人易患諸如精神分裂癥等重大精神疾病的遺傳因素是很有興趣的,因為我們知道,在精神分裂癥患者中,大約 80%的個體差異是由于遺傳差異造成的。人類基因組計劃和隨后的基因組科學革命為研究人類在基因組上的差異提供了工具。近年來使用的一種方法被稱為全基因組關聯研究(GWAS),其中每個個體的基因組在其基因組中的 100 萬個或更多的位置上進行特征化,以確定他們所擁有的遺傳密碼的字母(我們稱之為“變體”)。在那個地方。在確定了這些之后,研究人員在基因組的每個位置進行了統計測試,以確定被診斷為精神分裂癥的人在該位置是否或多或少有一個特定的變異。
讓我們想象一下,如果研究人員簡單地問,在每個地點的 P<;.05 測試是否有意義,而實際上在任何地點都沒有真正的效果,會發生什么。為此,我們從一個空分布生成大量的模擬 t 值,并詢問其中有多少在 p<;.05 處是重要的。讓我們這樣做很多次,每次都計算出有多少測試結果是重要的(參見圖[9.9](#fig:nullSim))。
```r
# simulate 1500 studies with 10,000 tests each, thresholded at p < .05
nRuns <- 1500 # number of simulated studies to run
nTests <- 10000 # number of simulated genes to test in each run
uncAlpha <- 0.05 # alpha level
uncOutcome <- replicate(nRuns, sum(rnorm(nTests) < qnorm(uncAlpha)))
sprintf("mean proportion of significant tests per run: %0.2f", mean(uncOutcome) / nTests)
```
```r
## [1] "mean proportion of significant tests per run: 0.05"
```
```r
# compute proportion of studies with at least one false positive result,
# known as the familywise error rate
sprintf("familywise error rate: %0.3f", mean(uncOutcome > 0))
```
```r
## [1] "familywise error rate: 1.000"
```
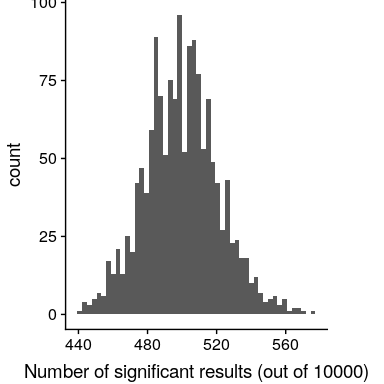
圖 9.9:每組 100 萬統計試驗中,當事實上沒有真實效果時,有效結果數量的柱狀圖。
這表明,所有測試中約有 5%在每次運行中都是顯著的,這意味著如果我們使用 p<;.05 作為統計顯著性的閾值,那么即使沒有真正顯著的關系存在,我們仍然會“找到”大約 500 個看似顯著的基因。NT(預期的重要結果數只是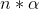)。這是因為當我們控制每個測試的錯誤時,我們沒有控制 _ 系列錯誤 _,或者所有測試中的錯誤,這是我們真正想要控制的,如果我們要查看大量測試的結果。使用 p<;.05,在上面的例子中,我們的家庭錯誤率是一個——也就是說,我們幾乎可以保證在任何特定的研究中至少出現一個錯誤。
控制家族誤差的一個簡單方法是將 alpha 水平除以測試次數;這被稱為 _bonferroni_ 修正,以意大利統計學家 Carlo Bonferroni 的名字命名。利用上述示例中的數據,我們在圖[9.10](#fig:bonferroniSim)中看到,只有約 5%的研究顯示使用校正后的α水平 0.000005 而不是名義水平 0.05 的任何顯著結果。我們已經有效地控制了家族誤差,這樣我們研究中的 _ 任何 _ 誤差發生的概率控制在 0.05 左右。
```r
# compute Bonferroni-corrected alpha
corAlpha <- 0.05 / nTests
corOutcome <- replicate(nRuns, sum(rnorm(nTests) < (qnorm(corAlpha))))
sprintf("corrected familywise error rate: %0.3f", mean(corOutcome > 0))
```
```r
## [1] "corrected familywise error rate: 0.046"
```
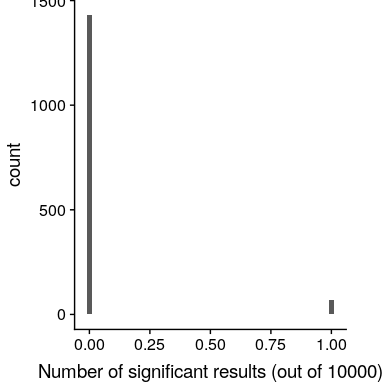
圖 9.10 在多次試驗中應用 Bonferroni 校正后,所有模擬運行的重要結果數量的柱狀圖。
- 前言
- 0.1 本書為什么存在?
- 0.2 你不是統計學家-我們為什么要聽你的?
- 0.3 為什么是 R?
- 0.4 數據的黃金時代
- 0.5 開源書籍
- 0.6 確認
- 1 引言
- 1.1 什么是統計思維?
- 1.2 統計數據能為我們做什么?
- 1.3 統計學的基本概念
- 1.4 因果關系與統計
- 1.5 閱讀建議
- 2 處理數據
- 2.1 什么是數據?
- 2.2 測量尺度
- 2.3 什么是良好的測量?
- 2.4 閱讀建議
- 3 概率
- 3.1 什么是概率?
- 3.2 我們如何確定概率?
- 3.3 概率分布
- 3.4 條件概率
- 3.5 根據數據計算條件概率
- 3.6 獨立性
- 3.7 逆轉條件概率:貝葉斯規則
- 3.8 數據學習
- 3.9 優勢比
- 3.10 概率是什么意思?
- 3.11 閱讀建議
- 4 匯總數據
- 4.1 為什么要總結數據?
- 4.2 使用表格匯總數據
- 4.3 分布的理想化表示
- 4.4 閱讀建議
- 5 將模型擬合到數據
- 5.1 什么是模型?
- 5.2 統計建模:示例
- 5.3 什么使模型“良好”?
- 5.4 模型是否太好?
- 5.5 最簡單的模型:平均值
- 5.6 模式
- 5.7 變異性:平均值與數據的擬合程度如何?
- 5.8 使用模擬了解統計數據
- 5.9 Z 分數
- 6 數據可視化
- 6.1 數據可視化如何拯救生命
- 6.2 繪圖解剖
- 6.3 使用 ggplot 在 R 中繪制
- 6.4 良好可視化原則
- 6.5 最大化數據/墨水比
- 6.6 避免圖表垃圾
- 6.7 避免數據失真
- 6.8 謊言因素
- 6.9 記住人的局限性
- 6.10 其他因素的修正
- 6.11 建議閱讀和視頻
- 7 取樣
- 7.1 我們如何取樣?
- 7.2 采樣誤差
- 7.3 平均值的標準誤差
- 7.4 中心極限定理
- 7.5 置信區間
- 7.6 閱讀建議
- 8 重新采樣和模擬
- 8.1 蒙特卡羅模擬
- 8.2 統計的隨機性
- 8.3 生成隨機數
- 8.4 使用蒙特卡羅模擬
- 8.5 使用模擬統計:引導程序
- 8.6 閱讀建議
- 9 假設檢驗
- 9.1 無效假設統計檢驗(NHST)
- 9.2 無效假設統計檢驗:一個例子
- 9.3 無效假設檢驗過程
- 9.4 現代環境下的 NHST:多重測試
- 9.5 閱讀建議
- 10 置信區間、效應大小和統計功率
- 10.1 置信區間
- 10.2 效果大小
- 10.3 統計能力
- 10.4 閱讀建議
- 11 貝葉斯統計
- 11.1 生成模型
- 11.2 貝葉斯定理與逆推理
- 11.3 進行貝葉斯估計
- 11.4 估計后驗分布
- 11.5 選擇優先權
- 11.6 貝葉斯假設檢驗
- 11.7 閱讀建議
- 12 分類關系建模
- 12.1 示例:糖果顏色
- 12.2 皮爾遜卡方檢驗
- 12.3 應急表及雙向試驗
- 12.4 標準化殘差
- 12.5 優勢比
- 12.6 貝葉斯系數
- 12.7 超出 2 x 2 表的分類分析
- 12.8 注意辛普森悖論
- 13 建模持續關系
- 13.1 一個例子:仇恨犯罪和收入不平等
- 13.2 收入不平等是否與仇恨犯罪有關?
- 13.3 協方差和相關性
- 13.4 相關性和因果關系
- 13.5 閱讀建議
- 14 一般線性模型
- 14.1 線性回歸
- 14.2 安裝更復雜的模型
- 14.3 變量之間的相互作用
- 14.4“預測”的真正含義是什么?
- 14.5 閱讀建議
- 15 比較方法
- 15.1 學生 T 考試
- 15.2 t 檢驗作為線性模型
- 15.3 平均差的貝葉斯因子
- 15.4 配對 t 檢驗
- 15.5 比較兩種以上的方法
- 16 統計建模過程:一個實例
- 16.1 統計建模過程
- 17 做重復性研究
- 17.1 我們認為科學應該如何運作
- 17.2 科學(有時)是如何工作的
- 17.3 科學中的再現性危機
- 17.4 有問題的研究實踐
- 17.5 進行重復性研究
- 17.6 進行重復性數據分析
- 17.7 結論:提高科學水平
- 17.8 閱讀建議
- References